본 매뉴얼은 리눅스(UBUNTU 20.04)환경에서 Terminal에 명령을 내리는 방식으로 작성되었습니다.
사용자는 UBUNTU 20.04 이상의 환경에서 리눅스 환경에 익숙해지는 과정을 거치면서 매뉴얼을 사용하시기 바랍니다.
https://sites.google.com/view/seoulqmt
University of Seoul QMT
Quantum materials theory at the University of Seoul
sites.google.com
Quantum Espresso 에서 코드를 돌릴 때에는 Input File 이라는 것이 있다. 우리가 원하는 분자구조에 대한 정보를 입력하는 것인데, 특별한 개발 환경이 필요하지 않고, vi, vim, gedit, notepad 등의 텍스트 에디터 프로그램에 입력하면 된다.
그렇다면, 어떤 파라미터들이 들어갈까? pw.x 라고 구글에 치면 다음과 같은 사이트가 나온다.

Input description. 이 사이트에 접속해 보면 다음과 같은 화면을 볼 수 있다.
이 화면에 나오는 하이퍼링크를 클릭하면 파라미터들에 대한 설명이 올라오게 된다.
이 파라미터들을 모두 사용하지는 않고, 필요에 따라 없애거나 다른 것으로 대체하기도 한다. 예를 들어, &System 아래 맨 처음에 나오는 ibrav 라는 변수는 분자 구조의 모양을 말해 주는 변수인데, 0,1,2,3,4,..등의 정수 코드를 입력받는다.
그런데, 이 변수를 0(사용자 설정에 따른다는 뜻)으로 입력한 뒤, CELL_PARAMETER 변수를 만들어 코드를 입력해서 모양을 직접 만들 수도 있다. 다른 ibrav 코드를 가지는 분자들의 모양도 이것으로 똑같이 만들 수 있다.
우선, MoS2를 계산할 때 사용하였던 input File을 보여 주겠다.
Linux Command 창에 vi MoS2_40.in 라고 친 뒤, 다음의 코드를 복사하여 붙여넣고 Input File을 하나 만들도록 하자.
그리고 pw.x는 아마 usr/bin위치에 있을 것이다. 이 프로그램을 Input file 위치에 두자.
| &control calculation = 'scf' #################################1 restart_mode='from_scratch' prefix='MoS2' ####################################2 etot_conv_thr = 1.0D-8 forc_conv_thr = 1.0D-4 tstress = .true. tprnfor = .true. pseudo_dir = '당신의 pseudo 폴더 디렉토리' ##########3 outdir='./tmp' / &system ibrav= 4 A=3.161 C = 20 nat= 3 ntyp= 2 ################4 ecutwfc = 40 ####################################5 occupations='smearing' smearing='gaussian' degauss=0.020000 / &electrons conv_thr = 1.0d-10 mixing_beta = 0.3 / ATOMIC_SPECIES Mo 95.94 Mo_ONCV_PBE-1.2.upf S 32.065 S_ONCV_PBE-1.2.upf ##########################6 ATOMIC_POSITIONS (crystal) Mo 0.666666619 0.333333314 0.750000039 S 0.333333360 0.666666714 0.627500011 S 0.333333360 0.666666714 0.872499989 ##########7 K_POINTS automatic 8 8 1 0 0 0 #######################################8 |
*Quautum Espresso 7.4 기준으로, '#' 가 주석으로 처리되지 않는 듯 합니다. ##가 있는 부분은 다 지우고 진행해 주세요.
1에 있는 ‘scf’는 수행될 작업의 종류를 말한다. 현재는 scf를 사용할 것이다. 나중에 relax, bands, vc-relax등의 다른 명령어들도 사용할 것이다. (vc : variable-cell)
2 에 있는 ‘Prefix’는 우리가 계산할 물질의 종류를 써 넣는 것이다.
3 은 다운받아 둔 PseudoPotential의 위치를 말한다. 이 위치를 정확히 입력하지 않으면 오류가 생긴다. 리눅스에서 포텐셜 파일이 있는 디렉토리를 찾아 가서 pwd를 입력하면 이 파일들의 위치가 나온다. 이것을 복사해서 붙여 넣으면 된다.
4 는 결정의 모양에 대한 정보들이다. ibrav는 앞서 설명하였듯이 모양을 말해 주는 것이고, A,C등은 맨 처음에 말했던 Lattice parameter 들이다. nat 은 number of atom 즉, 원자의 갯수, ntyp는 원자 종류의 갯수이다. MoS2는 원자 3개, 2개 종류의 원자이므로 각각 3, 2 를 입력한 것이다.
5는 파동함수에서 Kinetic Energy Cutoff (Ry) (Ry : atomic physics 에서 광자의 에너지, 파수가 리드버그 상수인 경우다. 1Ry=13.606eV) 를 말한다. 앞서 말한 potential energy cutoff와도 관련이 있다. 이 에너지를 하나하나 변화시켜서 output file 에서의 데이터를 변화시킬 것이다. 처음 시작은 40 정도로 해보도록 하자.
6 은 원자의 종류와 atomic weight, 그리고pseudo potential 파일 이름을 순서대로 적은 것이다. Mo와S원자를 각각 적어넣었다.
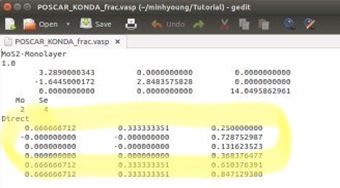
7은 원자들의 위치를 말한다. 이 위치는 앞서 만든 POSCAR파일을 열어보면 알 수 있다. Direct밑에 있는 숫자들을 복사해서 붙여 넣으면 된다. 만일 앞에서Fractional 이 아니라 Cartesian 으로 저장했다면 계산이 끝나도 잘못된 결과가 나올 확률이 높다.
8은 말 그대로 K-Point를 입력하는 곳이다.
K-Point라는것은 고체물리학에서 말하는 1st Brillouin zone(그러니까 각장 큰 주기성을 가지는 공간)내의 한 점을 말한다.
여기까지가 Input File에 대한 내용이다.
'DFT > Calculation' 카테고리의 다른 글
| 7. Total Energy 그래프 그리기 (2023.08.03 보충 설명 추가) (0) | 2020.09.07 |
|---|---|
| 6. Output File (0) | 2020.09.07 |
| 4. Pseudo Potential(2023.07.26수정 완료) (7) | 2020.09.02 |
| 3. Xcrysden 사용법 (2023.07.26수정 완료) (2) | 2020.08.31 |
| 2.VESTA 사용법 - Monolayer MoS2 만들기 (2023.08.03 수정) (5) | 2020.08.28 |



